Summary
Humanin is a small protein produced by the mitochondria, originally discovered in 2001 for its ability to protect brain cells from damage related to Alzheimer’s disease. Since then, it has been recognized as part of a larger group of mitochondrial-derived peptides that help cells survive under stress. Research has shown that Humanin plays a protective role in many tissues, including the brain, heart, blood vessels, and pancreas. It helps regulate metabolism, reduce inflammation, prevent cell death, and improve resilience to aging-related damage. Humanin levels decline with age, and lower levels are often seen in people with chronic diseases, while higher levels are found in long-lived individuals. Because of its broad protective effects and connection to longevity, Humanin is now being studied as a potential therapeutic tool to improve healthspan and reduce age-related decline.
Humanin Overview
Humanin (HN) is a 24–amino acid peptide encoded in the mitochondrial 16S ribosomal RNA gene (MT-RNR2) and was first identified in 2001 as a neuroprotective “rescue factor” that blocks neuronal death caused by Alzheimer’s disease (AD)-related insults. Subsequent studies revealed HN as a paradigm for mitochondrial-derived peptides with pleiotropic cytoprotective actions. HN exists in both intracellular and secreted forms, engaging multiple signal transduction pathways to promote cell survival. It binds pro-apoptotic proteins (e.g. Bax, Bid) to halt mitochondrial cell-death cascades and interacts with specific cell-surface receptors to activate pro-survival signaling (JAK/STAT3, PI3K/AKT, ERK1/2). In diverse in vitro and animal models, HN and more potent analogues (such as S14G-humanin) protect neurons, pancreatic β-cells, cardiomyocytes, endothelial cells and other cell types from oxidative stress, metabolic insults, and apoptotic injury. Moreover, HN improves physiological function in models of AD, atherosclerosis, diabetes, and aging, including extension of lifespan in C. elegans. These multifaceted benefits position HN as a compelling subject in basic and translational science. This review critically examines the current evidence on HN’s molecular mechanisms, preclinical efficacy, and unresolved questions, highlighting HN’s emerging role as a mitochondrial signal peptide with broad cytoprotective potential.
Molecular Origin & Structure
Humanin is encoded by a 75-base pair open reading frame within the mitochondrial 16S rRNA gene (MT-RNR2), making it a rare example of a “gene within a gene” in the mitochondrial genome. First identified by Hashimoto et al. during a screen for neuroprotective cDNAs in Alzheimer’s brain tissue, the resulting peptide is 24 amino acids long (MAPRGFSCLLLLTSEIDLPVKRRA), though mitochondrial translation may start with formyl- methionine. Humanin shares 92–100% sequence homology with 13 nuclear MTRNR2L loci, thought to be nuclear mitochondrial pseudogenes (Numts). Orthologs exist across species—for instance, “rattin” in rats is a 38-residue variant with a C-terminal extension—suggesting strong evolutionary conservation.
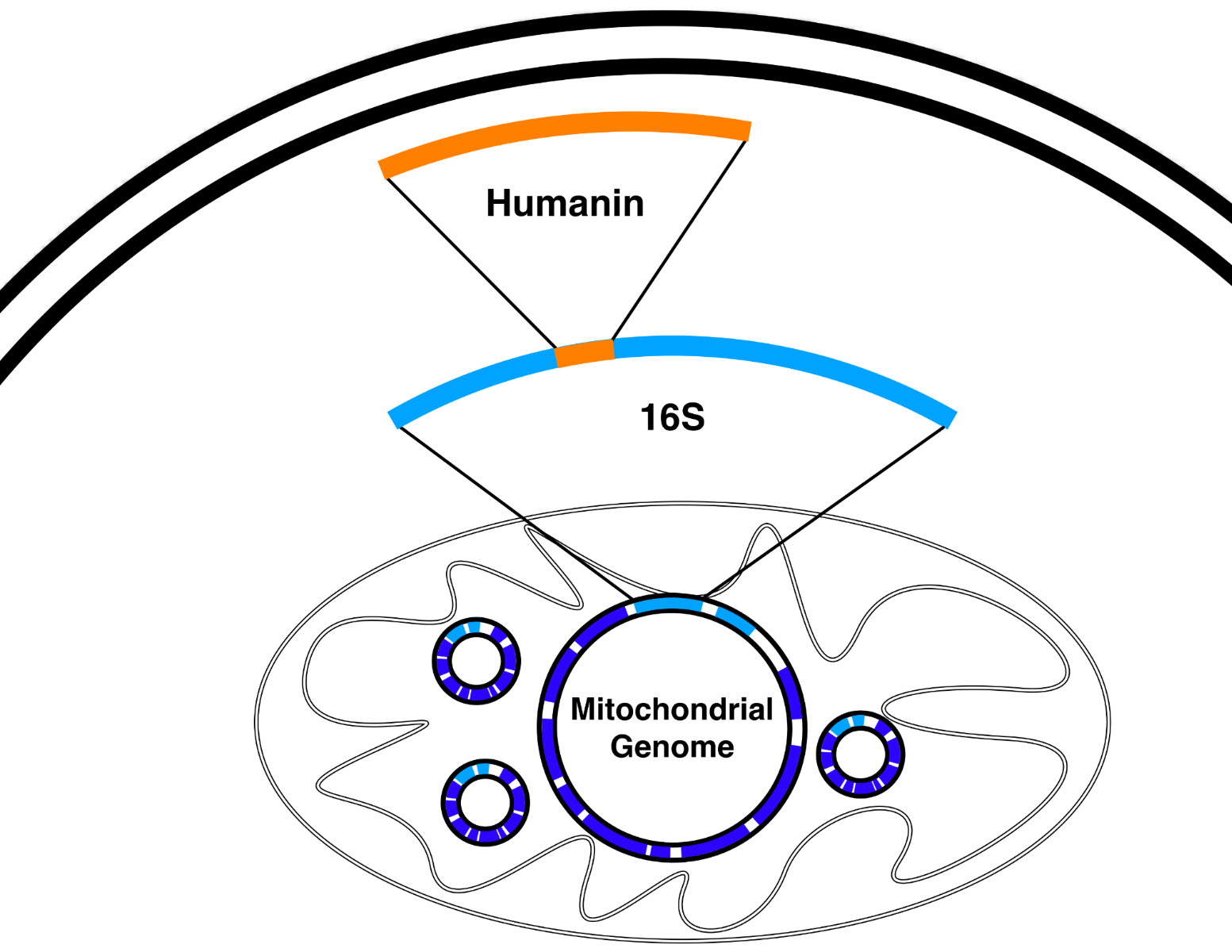
Sequence Variants and Analogs:
Structure–function analyses have identified critical residues in the Pro³–Ser¹⁴ core domain. Mutations at Ser⁷, Cys⁸, Leu⁹, Leu¹², Thr¹³, Ser¹⁴, or Pro¹⁹ abolish bioactivity. Replacing Ser¹⁴ with glycine produces HNG, a potent analogue that shows >1000-fold higher cytoprotective activity, likely due to greater conformational flexibility and target binding affinity. Other notable analogues include HNGF6A (S14G with Phe⁶→Ala), which enhances insulin secretion without binding IGFBP-3, and Colivelin, a chimeric peptide merging a truncated HNG with a neurotrophic factor segment that shows femtomolar efficacy in memory models. Recent work has yielded stabilized analogs—like cyclic D-Ser14 humanin (cHND14) and “HUJ” analogs—with enhanced resistance to proteolysis and potent protection in models of ischemia and excitotoxicity.
Secretion and Localization:
Despite lacking a classical signal sequence, HN is secreted via a non-canonical mechanism. Yamagishi et al. identified two hydrophobic motifs (Leu⁹–Leu¹¹ and Pro¹⁹–Val²⁰) essential for secretion; mutating Leu10 abolished extracellular release without affecting intracellular levels. These motifs may serve as an internal secretion signal, facilitating export through unconventional pathways such as direct membrane translocation or vesicular trafficking. HN appears to localize first to the mitochondrial matrix or inner membrane before being released into the cytosol and then extracellular space. Dimerization, also governed by key residues like Ser7 and Leu9, correlates with enhanced activity, potentially by enabling high-avidity receptor interactions.
Stability and Expression Dynamics:
Intracellular HN is regulated by the ubiquitin–proteasome system. TRIM11, an E3 ubiquitin ligase, binds HN and promotes its degradation; its absence elevates HN levels, while proteasome inhibitors block turnover. MT-RNR2 mRNA is highly expressed in metabolically active tissues—heart, skeletal muscle, kidney, liver, testis—and HN peptide is detectable in these tissues as well as in plasma and cerebrospinal fluid. HN levels decline with age in rodents and humans, a trend thought to reflect diminishing cellular resilience with aging. These patterns suggest a regulated system in which HN acts as a mitochondria-derived signal peptide, coordinating stress responses across cellular compartments.
Preclinical Evidence — In Vivo (Animal Models)
Neurodegeneration Models:
HN’s neuroprotective effects have been demonstrated across a variety of rodent neurodegeneration models. In triple-transgenic Alzheimer’s mice (mutant APP, PSEN1, Tau), intranasal HNG over 3 months reduced Aβ plaque burden, lowered glial inflammation, and improved maze performance. APP/PS1 mice treated peripherally with HNG also showed diminished brain amyloid load and cognitive recovery, indicating CNS penetration and functional rescue. In acute models, HNG blocked memory loss induced by Aβ_25–35 peptide and preserved performance in scopolamine-induced cholinergic impairment. HN has also countered non-amyloid insults: in rats, it protected neurons from toxicity, and in polyglutamine neurodegeneration models, HN reduced protein aggregates and ASK1-driven neuronal death. However, effects are not universal; in one Huntington’s disease model, HN showed no benefit, suggesting disease-specific variables modulate its action.
In ischemic stroke models, intracerebroventricular HNG post-MCAO reduced infarct size by ~50%, decreased apoptosis, and improved neurological scores. Mechanistically, HNG suppressed ERK and PARP activation while boosting AKT, with PI3K inhibition abolishing its effect. Combined HNG and Necrostatin-1 (a necroptosis inhibitor) yielded synergistic benefits—~80% infarct reduction and greater functional recovery versus monotherapy. In vitro oxygen-glucose deprivation studies confirmed enhanced dual-pathway protection. In hemorrhagic stroke, systemic HNG reduces brain edema and neuronal death while facilitating mitochondrial transfer from astrocytes to neurons—a possible endogenous repair response.
HN also shows promise in Parkinson’s disease models. In MPTP-exposed mice, peripheral HN treatment preserved motor function, dopaminergic neurons, and reduced oxidative stress in the striatum. Though early, these data support HN’s broader neuroprotective potential via anti-apoptotic and mitochondrial-preserving mechanisms.
Metabolic Regulation:
HN modulates metabolism in rodent models, particularly insulin sensitivity and glucose homeostasis. Central HN delivery enhanced insulin action during hyper insulinemic–euglycemic clamps, primarily through suppressed hepatic glucose output and increased muscle uptake. This effect depended on hypothalamic STAT3 activation. Peripherally, intravenous HNGF6A improved glucose disposal and reduced gluconeogenesis in diabetic rats. Chronically, mid-life HNG administration improved glucose tolerance, reduced inflammatory cytokines, and lowered adiposity. HN also supports pancreatic β-cell integrity: in streptozotocin-treated mice, HNG preserved islet architecture and delayed hyperglycemia onset. Transgenic mice overexpressing HN were more insulin sensitive and resistant to diet-induced insulin resistance. Long-lived Ames dwarf mice exhibit high HN levels, while short-lived GH-transgenic mice have low levels, paralleling favorable metabolic traits (low insulin, high sensitivity). Collectively, these models position HN as a metabolic regulator with anti-diabetic potential.
Cardioprotection and Vasoprotection:
HN exerts protective effects in the cardiovascular system. In myocardial ischemia/reperfusion models, HNG reduced infarct size by over 40%, improved ejection fraction and cardiac output, and diminished cardiomyocyte apoptosis. HN expression increased in ischemic tissue, and human coronary plaques showed HN immunoreactivity—hinting at an adaptive stress response. In ApoE⁻/⁻ mice, HNGF6A treatment preserved endothelial function, halved aortic plaque area, and reduced oxidative stress markers in lesions. It also protected against microvascular damage in the kidneys, suggesting end-organ benefits. Corroborating this, patients with endothelial dysfunction have lower circulating HN and reduced flow-mediated dilation. HN also limited vascular remodeling: in rat carotid injury, it reduced smooth muscle proliferation and inflammatory signaling. Altogether, these findings underscore HN’s role in vascular homeostasis and ischemic resilience.
Lifespan and Healthspan:
HN is linked to longevity in several models. In C. elegans, HN expression extended lifespan in a DAF-16-dependent manner and conferred stress resistance. In mammals, HNG- treated mice maintained superior glucose metabolism, lower cytokine levels, and increased late- life activity. Offspring of human centenarians’ exhibit ~30% higher plasma HN, while patients with age-related diseases (e.g., AD, MELAS) show reduced levels. Notably, naked mole rats—remarkably long-lived rodents—maintain youthful HN levels into old age, unlike typical rodents. While elevated HN has been detected in some cancers (see later section), in aging models, HN consistently delays decline without observed trade-offs. These findings suggest that persistent HN expression enhances cellular resilience, supporting its candidacy as a longevity- associated factor in geroscience research.
Comparative & Synergistic Biology
Humanin (HN) is the first discovered member of a now-expanding class of mitochondrial- derived peptides (MDPs), short peptides encoded within mitochondrial rRNA loci that exert cytoprotective effects. Among the most studied MDPs alongside HN are the small humanin-like peptides (SHLPs 1–6) and MOTS-c, each with distinct but sometimes overlapping roles in stress adaptation.
The SHLPs were identified from ORFs within the 16S rRNA region that also encodes HN. SHLP2 and SHLP3, for instance, show pro-survival, anti-apoptotic functions in vitro, with SHLP3 improving glucose tolerance and reducing adiposity in vivo. Conversely, SHLP6 exhibits pro-apoptotic effects, possibly serving as a counter-regulatory factor that induces mitophagy or inhibits unchecked proliferation. These findings suggest SHLPs form a network with both synergistic and antagonistic actions, modulating cell fate under stress.
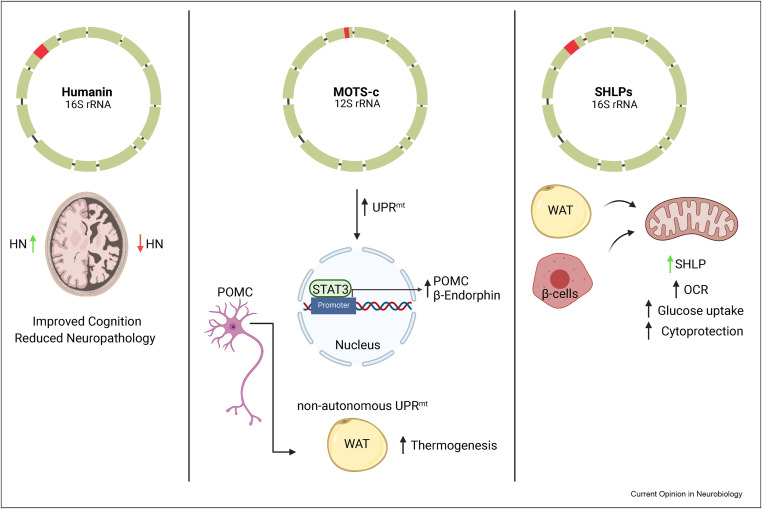
MOTS-c, a 16-amino acid peptide encoded in the mitochondrial 12S rRNA, complements HN’s actions. It translocates to the nucleus during metabolic stress and activates genes involved in homeostasis via AMPK. Like HN, MOTS-c enhances insulin sensitivity and protects against diet-induced obesity in mice. Notably, both peptides decline with age and are elevated in long-lived models, implying that MDPs collectively sustain stress resistance across tissues throughout youth, waning with aging. Given these overlaps, combined interventions involving HN are being explored. A clear example is HN (HNG) co-administered with the necroptosis inhibitor Necrostatin-1 in stroke models, where simultaneous targeting of apoptotic and necrotic pathways resulted in superior neuroprotection. Similar additive effects have been reported with anti-oxidants or neurotrophic peptides. Colivelin, a hybrid of HN and a fragment of activity-dependent neurotrophic factor (ADNF), outperformed HN alone in memory rescue and ischemia protection models. Another study combining HN with a peptide derived from an integrin-binding domain (HV) showed enhanced neural adhesion and survival compared to either alone.
The interplay between HN and other MDPs can also be regulatory. VSTM2L, a secreted protein, binds and antagonizes both HN and MOTS-c, suggesting coordinated regulation of their availability. Their expression appears to be temporally aligned—MOTS-c rises early in response to metabolic stress to modulate cellular energetics, while HN follows to limit apoptotic damage. Exercise exemplifies this relationship: endurance training elevates both peptides in muscle, correlating with improved insulin sensitivity and reduced apoptosis. In mouse models, combining exercise with MDP analogs enhanced metabolic health more than either intervention alone.
Such synergy has spurred interest in pairing HN analogs with caloric restriction mimetics or IGF-1 modulators, given HN’s influence on the insulin/IGF axis. Additionally, HN analogs are being evaluated as adjuvants in cancer therapy to reduce chemotherapy toxicity. In preclinical studies, HNG preserved bone and germ cell integrity during cisplatin treatment without shielding tumor tissue. However, this protective potential also raises concerns: HN could, under some conditions, aid tumor survival, making dosage and context critical variables. Some reports suggest HN may sensitize tumors to TNF-α, while others associate elevated HN with malignancy.
Overall, HN functions not in isolation but as part of a broader signaling ensemble. Its interactions with MDPs and exogenous agents position it as a keystone in adaptive responses to cellular stress. However, the presence of antagonists like SHLP6 and VSTM2L underscores the importance of understanding the timing, tissue-specificity, and balance among mitochondrial peptides. Future studies should continue to map this network, define synergy and redundancy, and determine how HN-based strategies might best be integrated into multifactorial approaches to enhance stress resilience and prevent degenerative decline.
Conclusion
Humanin (HN) exemplifies the power of mitochondria-encoded microproteins in regulating cell fate. Initially discovered for its neuroprotective effects in Alzheimer’s models, HN is now recognized as a broadly cytoprotective, pleiotropic peptide. Its dual mechanism—blocking apoptosis intracellularly and activating survival signaling extracellularly—allows it to counteract key drivers of aging such as oxidative stress, protein aggregation, and metabolic dysfunction. Across diverse preclinical models, HN and its analogues have repeatedly preserved tissue viability, from neurons and cardiomyocytes to pancreatic β-cells. Its integration into a broader network of mitochondrial-derived peptides (MDPs) underscores its role as part of an intrinsic cellular defense system.
While promising, HN biology is not without complexities. Its context-specific effects, potential interactions in cancer models, and unresolved mechanistic questions reflect the nuanced reality of mitochondrial signaling. Nevertheless, the weight of evidence favors HN as a net positive modulator of cellular resilience. As a therapeutic candidate, HN stands out for its multi-targeted effects an advantage in treating complex, multifactorial diseases like Alzheimer’s, myocardial infarction, and metabolic syndrome. Rather than acting on a single receptor or pathway, HN shifts cells into a survival-promoting state across multiple axes, offering a systems-level approach to protection.
The challenge ahead is delivery. Native HN is short-lived, and optimizing stability, potency, and tissue targeting will be critical. Potent analogues, or gene therapy approaches that locally enhance HN expression, represent the most viable paths forward. If successful, HN- based interventions could redefine treatment strategies for age-related diseases and potentially extend healthspan.
More broadly, HN’s discovery represents a shift in how we view the mitochondrial genome, from a static catalog of respiratory genes to a dynamic source of regulatory peptides. Humanin helped open the door to this new field, revealing that mitochondria are not just energy producers but central players in stress signaling and intercellular communication. The path forward will require advanced tools, careful study design, and rigorous validation. But the foundation is already strong.
In short, Humanin is no longer just an Alzheimer’s curiosity, it is now a leading candidate in geroscience, with far-reaching implications for how we understand and intervene in the aging process. As research continues, HN may become one of the first mitochondrial peptides to transition from molecular discovery to practical intervention in age-related disease. Its future lies not only in what it can do alone, but how it can be integrated into the broader orchestration of cellular longevity mechanisms.
References
- Hashimoto Y., Niikura T., Tajima H., et al. A rescue factor abolishing neuronal cell death by a wide spectrum of familial Alzheimer’s disease genes and Aβ. Proc Natl Acad Sci USA. 2001;98(11):6336–6341.
- Hashimoto Y., Niikura T., Ito Y., et al. Detailed characterization of neuroprotection by a rescue factor Humanin against various Alzheimer’s disease-relevant insults. J Neurosci. 2001;21(23):9235–9245.
- Guo B., Zhai D., Cabezas E., et al. Humanin peptide suppresses apoptosis by interfering with Bax activation. Nature. 2003;423(6938):456–461.
- Ikonen M., Liu B., Hashimoto Y., et al. Interaction between the Alzheimer’s survival peptide Humanin and insulin-like growth factor-binding protein 3 regulates cell survival and apoptosis. Proc Natl Acad Sci USA. 2003;100(22):13042–13047.
- Cobb L.J., Lee C., Xiao J., et al. Naturally occurring mitochondrial-derived peptides are age-dependent regulators of apoptosis, insulin sensitivity, and inflammatory markers. Aging (Albany NY). 2016;8(4):796–809.
- Yamagishi Y., Hashimoto Y., Niikura T., Nishimoto I. Identification of essential amino acids in Humanin, a neuroprotective factor against Alzheimer’s disease-relevant insults. Peptides. 2003;24(4):585–595.
- Terashita K., Hashimoto Y., Niikura T., et al. Two serine residues distinctly regulate the rescue function of Humanin, an inhibiting factor of Alzheimer’s disease-related neurotoxicity: functional potentiation by isomerization and dimerization. J Neurochem. 2003;85(6):1521–1534.
- Muzumdar R.H., Huffman D.M., Calvert J.W., et al. Acute humanin therapy attenuates myocardial ischemia and reperfusion injury in mice. Arterioscler Thromb Vasc Biol. 2010;30(10):1940–1948.
- Chiba T., Yamada M., Hashimoto Y., et al. Development of a femtomolar-acting Humanin derivative named colivelin by attaching activity-dependent neurotrophic factor to its N-terminus: characterization of colivelin-mediated neuroprotection against Alzheimer’s disease-relevant insults in vitro and in vivo. J Neurosci. 2005;25(52):10252–10261.
- Gilon C., Gitlin-Domagalska A., Lazarovici P., et al. Novel Humanin analogs confer neuroprotection in vitro and in vivo by mutations that stabilize the peptide in an active conformation. Neuropharmacology. 2020;168:108013.
- Wang J., Wang Y., Lin H., et al. Humanin, a mitochondrial-derived peptide, is secreted by astrocytes and improves neuronal energy metabolism. Cell Metab. 2021;33(10):2077–2093.e9.
- Niikura T., Kita Y., Abe Y., et al. A tripartite motif protein TRIM11 binds and destabilizes Humanin, a neuroprotective peptide against Alzheimer’s disease-relevant insults. Eur J Neurosci. 2003;17(6):1150–1158.
- Muzumdar R.H., Huffman D.M., Atzmon G., et al. Humanin: a novel central regulator of peripheral insulin action. PLoS ONE. 2009;4(7):e6334.
- Ying G., Iribarren P., Zhou Y., et al. Humanin, a newly identified neuroprotective factor, uses the G-protein-coupled formylpeptide receptor-like-1 (FPRL1) as a functional receptor. J Exp Med. 2004;199(5):621–627.
- Hashimoto Y., Kurita M., Aiso S., Nishimoto I., Matsuoka M. Humanin inhibits neuronal cell death by interacting with a cytokine receptor complex or complexes involving CNTF receptor α/WSX-1/gp130. Mol Biol Cell. 2009;20(12):2864–2873.
- Jia Y., Yang L., Chen Q., et al. Humanin analog protects against germ cell apoptosis and heat-induced testicular injury via the IL-27 receptor (WSX-1)/gp130/STAT3 pathway. Cell Death Dis. 2021;12(7):686.
- Matsuoka M., Hashimoto Y. Humanin and the receptors for Humanin. Mol Neurobiol. 2010;41(1):22–28. 18. Ma Z., Liu Z. Humanin decreases mitochondrial membrane permeability by interacting with Bax and Bid. Biochim Biophys Acta. 2018;1859(9):789–801.
Product available for research use only: